
Introduction
The FDA recently published a set of 5.6 million medical device reports previously hidden from the public. These “Alternative Summary Reports” (ASR) constitute over 40% of the total device reports now available.
The FDA characterized data from these hidden reports as containing “well-known and well-established risks” with the implication that the information within was of limited value. Our analysis suggests the opposite
One of the Food and Drug Administration’s core responsibilities is to collect and publish reports describing medical device problems and adverse events. This real-world data can help inform patients, doctors, and researchers as to potential risks to patient health, and weigh the comparative cost-benefit tradeoffs of similar devices.
The MAUDE (Manufacturer and User Facility Device Experience) database is the FDA’s largest repository of medical device reports. Regular, publicly released updates provide information on thousands of medical device product classes.
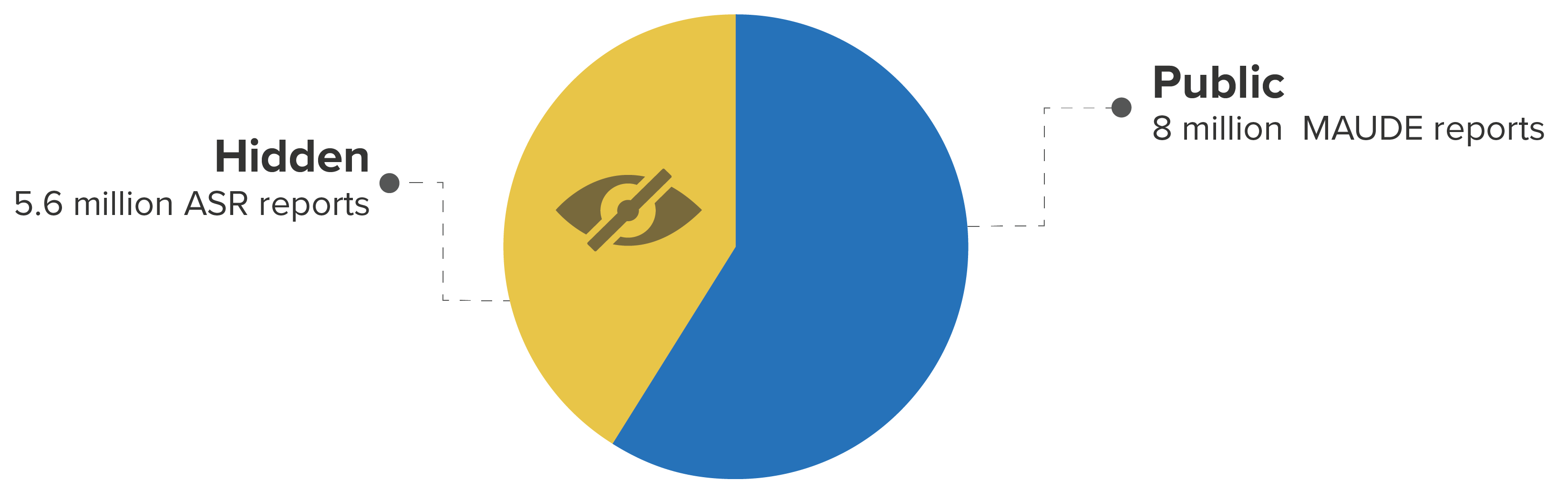
Breakdown of public/private FDA device reports between 1997 and 2019
- The sheer volume of hidden data, and narrow overlap with publicly reported issues, severely limits discovery of patient health-related patterns.
- Information in the hidden reports is more likely to be associated with life-threatening devices and to contain potentially serious problems.
- Reports for different manufacturers were hidden at varying rates (even among devices within the same product code), creating a skewed impression of which devices experienced the most problems.
The sheer volume of hidden data, and narrow overlap in reported issues, severely limits discovery of patient health-related patterns.
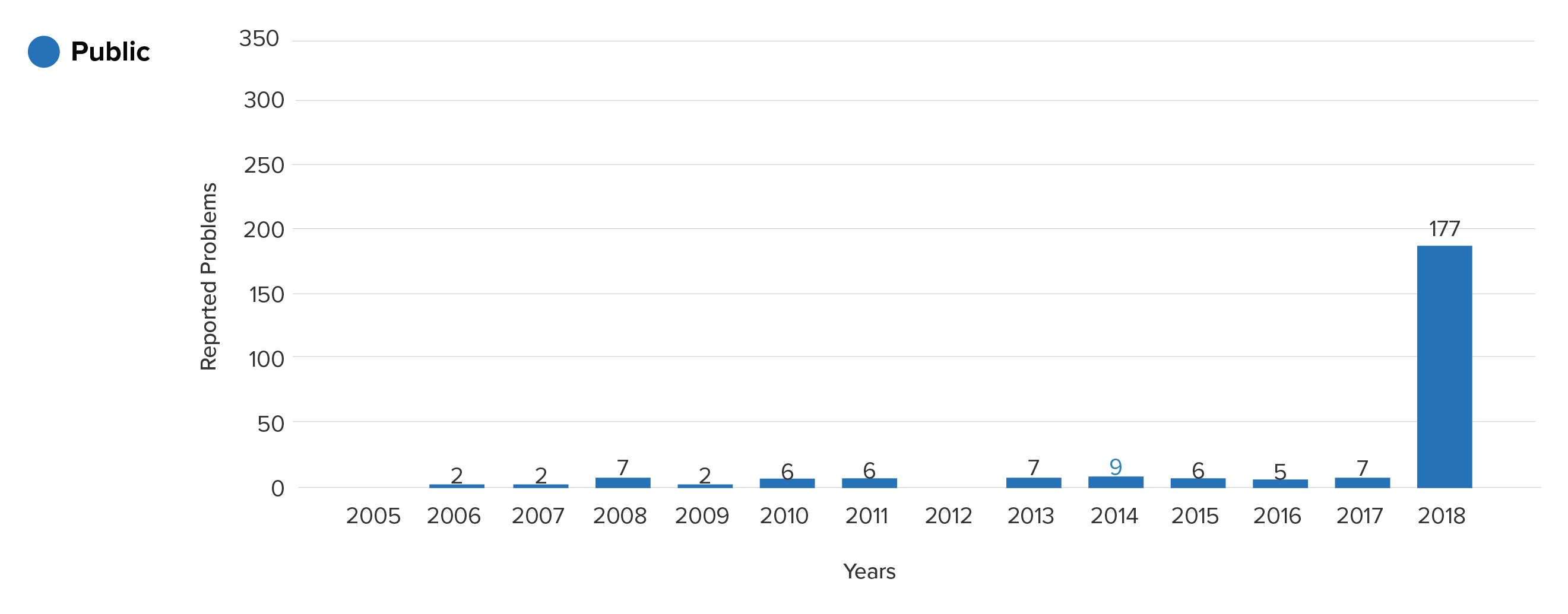
Suppose a hospital is using an implantable port device (product code LJT) to take blood and would like to know the frequency of device breakage.
This is the yearly trend seen when considering the public data only:
Device breakage reports for product class LJT from the MAUDE (public)
Prior to 2017, the logical conclusion would be that these devices rarely have problems, and hence pose minimal risk. Noticing the large spike in 2018, one may have wondered whether a change in manufacturing protocols suddenly caused a whole class of devices to experience serious problems.
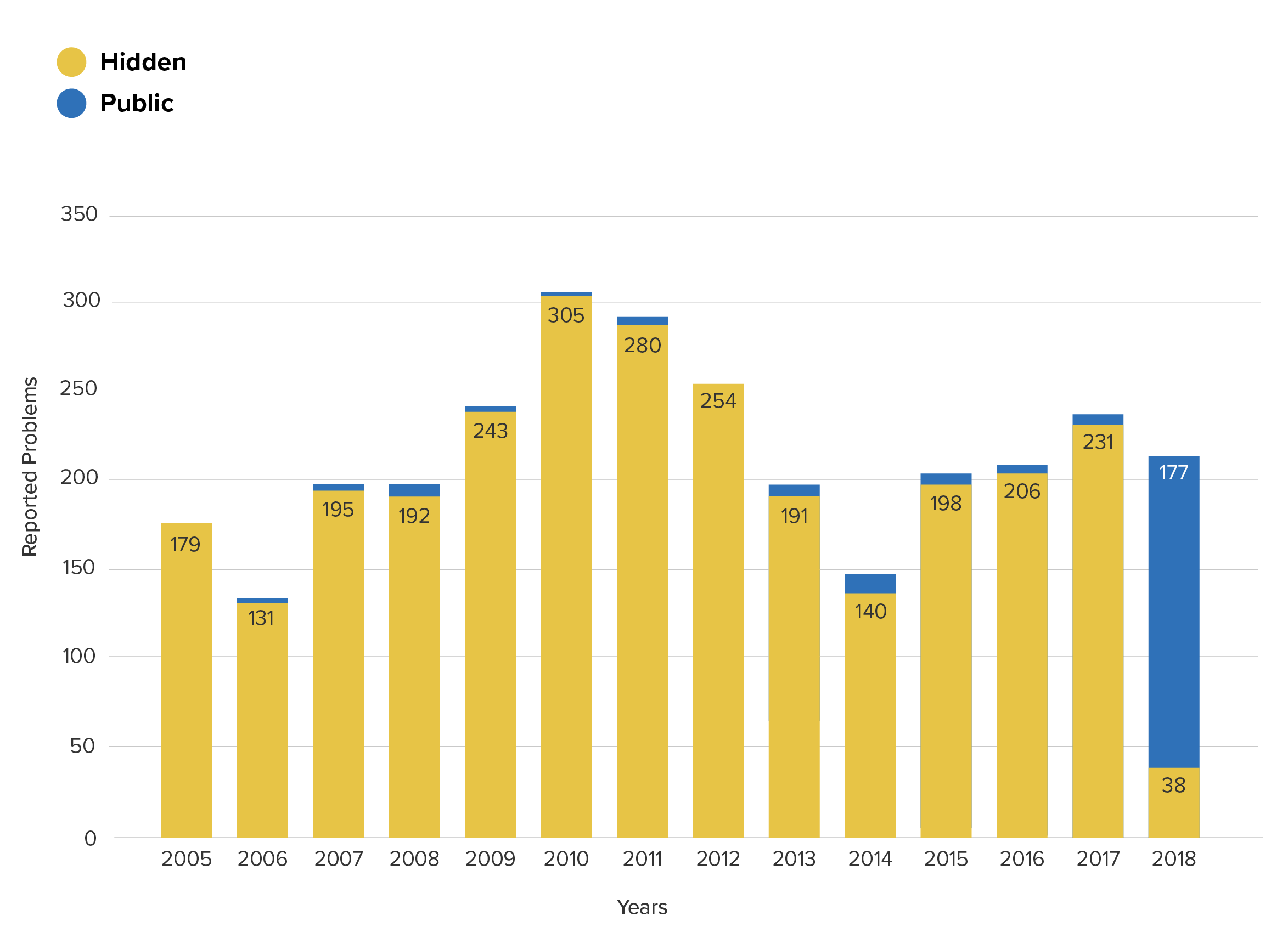
Now consider the yearly trend once the private data is added in:
Device breakage reports for product class LJT from the MAUDE (public) and ASR (hidden) databases
With the benefit of the extra data, both conclusions now seem clearly wrong. Prior to 2017, the frequency of these problems was an astonishing 47 times higher than was indicated by the reports which were published in MAUDE, and the story of 2018 becomes one of continuity – not of difference.
Information in the hidden reports are more likely to be associated with life-threatening devices and contain potentially serious problems.
102 (out of roughly 6500) medical device product classes had hidden reports. On average, over 60% of reports for these classes were hidden. These 102 classes include Implantable Pacemaker Pulse-Generators (DXY), Intraocular Lens (HQL), and Heart-Valve Tissue (LWR) and other high-risk devices. Overall, according to the FDA’s own classification, the hidden report database contains a much higher percentage of both implanted and life-sustaining devices. 44% of the hidden device classes are implantable and 14% are life-sustaining compared to 10% implantable and 3% life-sustaining in the public database.
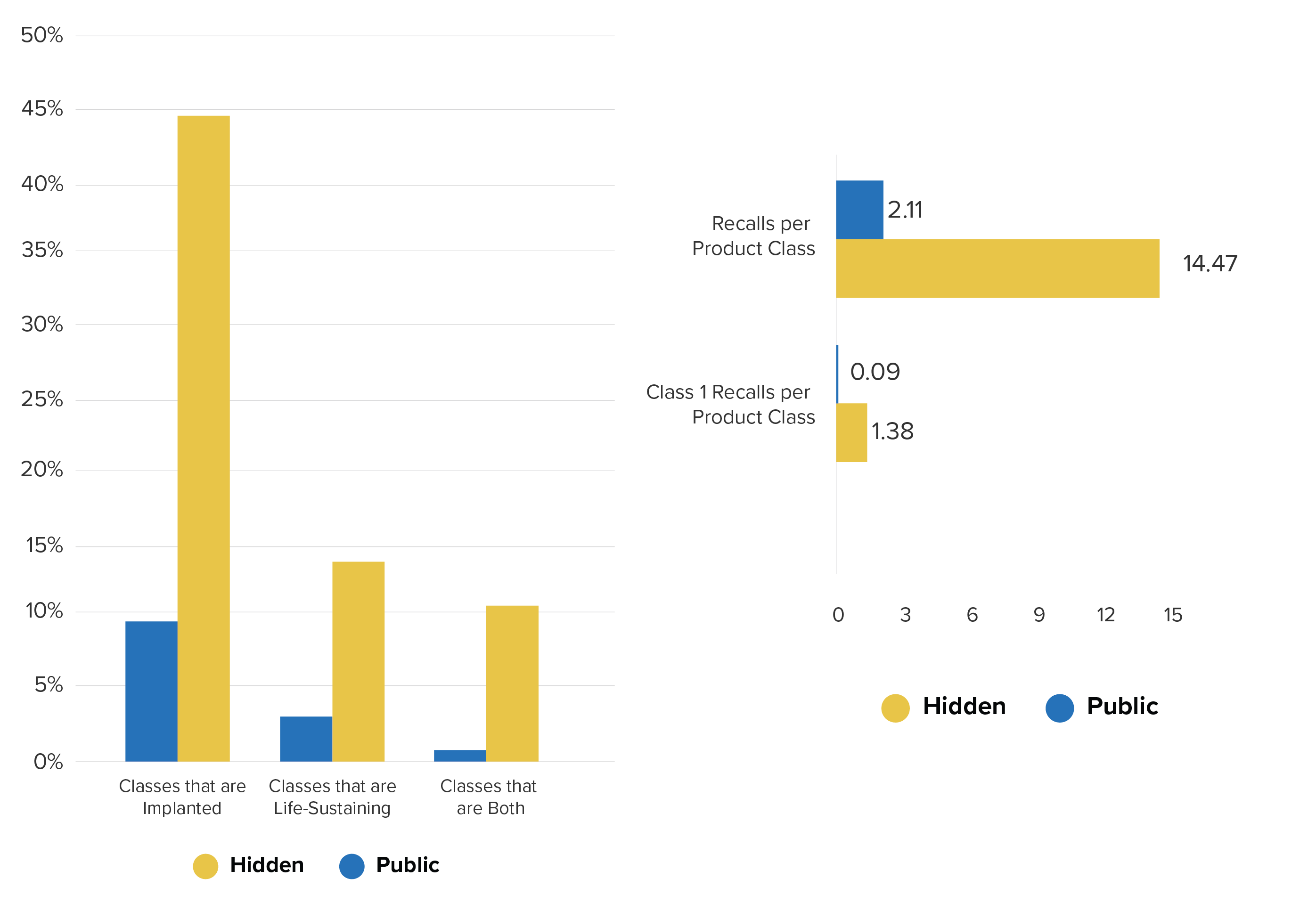
Product class percentages by class categories (left), reports per product class — all recalls and Class 1 recalls only (right)
Moreover, the device classes with hidden reports not only contain patient safety-critical products, but a greater ratio of Class I (life-threatening) recalls when compared to all device classes in the public database.
This meant that crucial information was shielded from the public about devices whose problems could lead to much more serious adverse events and life-threatening recalls. It also means that doctors were not able to assess device risk in real time because so many reports were hidden. A recall may have been the first public indication of significant device risk.
The reports were hidden at different rates for different manufacturers, creating a skewed impression of which devices were having the most problems
Even within the same product code, there are wide variations in individual manufacturer representation. For example, the class of Intra-Aortic Balloon Pumps (product code DSP) has a total of 34,384 hidden and 16,207 public reports. Among the manufacturers of these devices, Maquet (a subsidiary of Getinge) had almost 80% of its total reports hidden, while all Teleflex Reports were public.
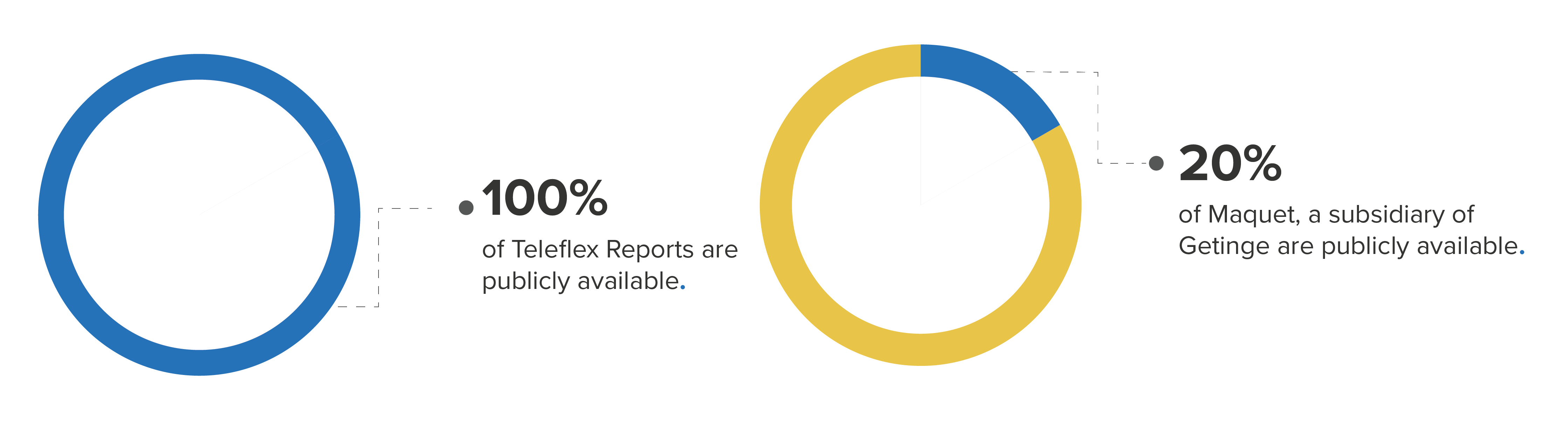
Percentage of public reports for product class DSP by manufacturer (Getinge and Teleflex)
These hidden reports are far from innocuous, describing serious product problems such as balloon leaks, ruptures and device alarm issues.
These issues were the subject of a series of 21 recalls in the past 10 years. Of these, seven were Class I recalls, a classification reserved for life-threatening issues.
What’s disturbing is that risk signals were present long before Getinge finally recalled their products, lurking in the hidden reports.
In 2017 and 2018 there were a series of Class 1 recalls for the CS100 and CS300 Devices by Getinge. Device problems associated with these recent recalls included electrical, autofill and pump failure, the very same problems that had been clear in the hidden reports. For example, a Getinge Class I recall reads:
“The device failed to pump due to an electrical test failure code #58 (power up vent test fail), maintenance code #3, and an autofill failure which has been associated to a patient death due to the failure of the device to initiate therapy.”

Getinge Class 1 recalls for DSP product class

Hidden (ASR) reported problems related to recall issues for DSP
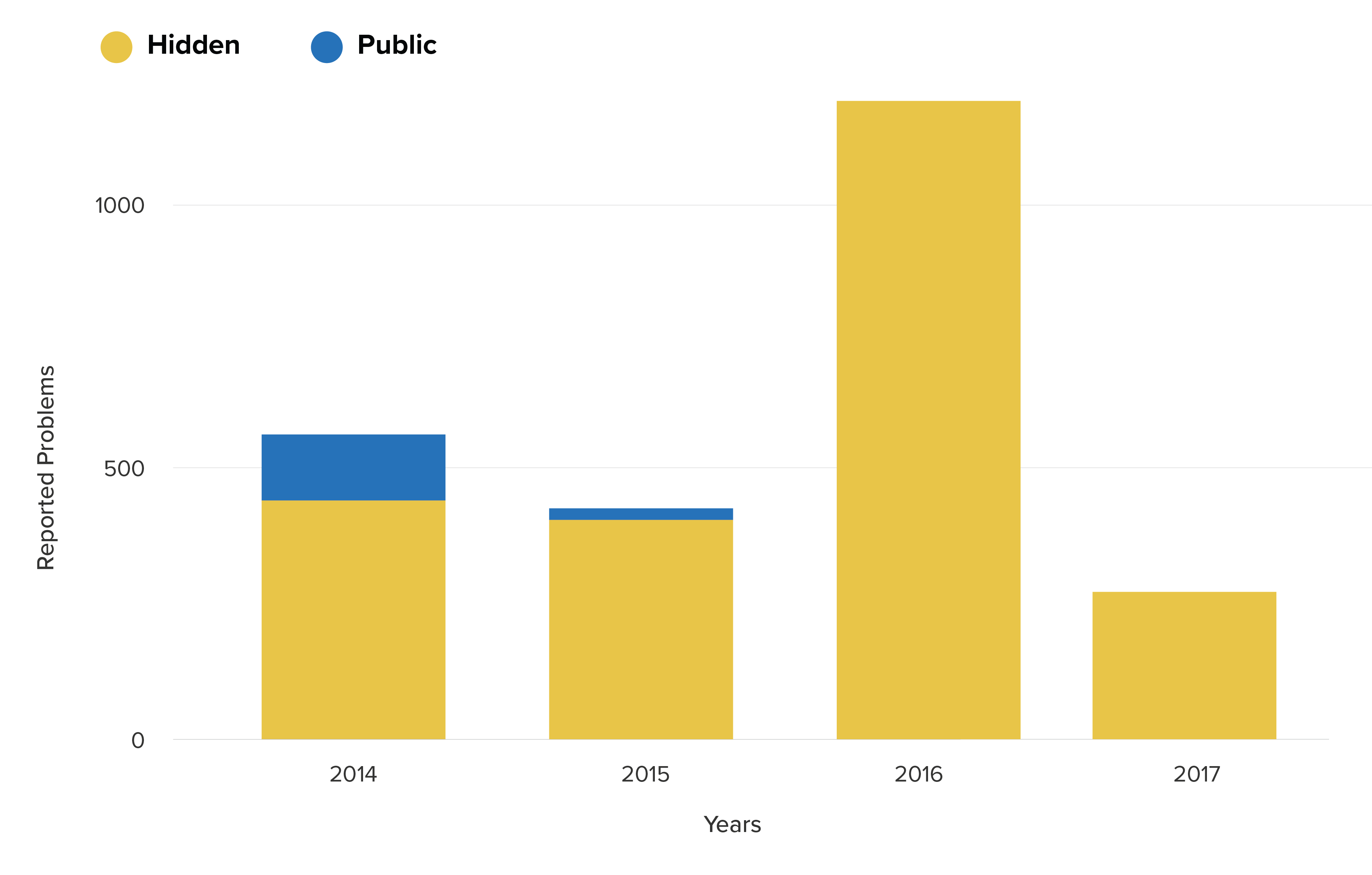
Yearly trend for these problems for the ASR (hidden) and MAUDE (public) databases
The timing is particularly unfortunate for patients using the Maquet devices through 2016. During this time, the FDA possessed a growing body of data suggesting significant product problems that they did not release to the public. Only in 2017 were the first related Class I recalls issued.
Conclusion
We cannot emphasize how important it is to patient safety for the FDA to publicly release as much data as possible, as quickly as possible. While the FDA’s motivation for not releasing 40% of device reports for so long is unclear, it’s reasonable to assume that had this data been accessible to the public, troubling patterns would have come to light much sooner, likely saving lives.
A wide variety of stakeholders may have been adversely impacted by the FDA’s decision not to release the hidden (ASR) data. These include patients, hospital purchasing agents, and device companies who may have been made to look worse than their peers — while actually having a better safety record.
Please feel free to reach out if you’d like to further discuss the issues and data sets presented in this white paper. We’d be happy to work with you.