
Having it all: Breaking through the safety/innovation trade-off with real-world data
A core part of the FDA’s job requires balancing the tension between streamlining medical product innovation, and patient safety.
To encourage faster innovation, and in response to the 21st Century Cures Act, the FDA recently revised the 510(k) application process to reduce regulatory burden. Now more Class II (described by the FDA as presenting “a moderate risk of harm”) medical devices can bypass the 510(k) regulatory process and go directly to market. They join over 2,100 Class I (“minimal potential for harm”) and Class II product classes previously added to the Premarket Exemption list.
Broadly speaking, we think reducing this regulatory burden is a good thing. Advanced analytics have matured to the point where real-world evidence (RWE) provides reliable risk signals, and proper monitoring allows for a much more robust, practical, long term approach to patient safety than the current 510(k) process (510k process hides liability landmines)
At the same time however, the FDA has unfortunately also eased post-market reporting requirements. As a replacement for the Alternative Summary Reporting (ASR) program (discontinued in 2019), in which selected manufacturers were granted exemptions from reporting device events in the public MAUDE database in favor of a hidden database (Hidden from View), companies can now use the Voluntary Malfunction Summary Reporting (VMSR) program. This is not much of an improvement. We note three areas of concern.
- The VMSR allows thousands of product classes to submit summary reports, rather than full reports. This makes it a much less rich primary source for assessing product safety.
- It allows manufacturers to provide only a single report summarizing the experience of thousands of patients. Important nuances involving a subset of the cases are frequently omitted, making pattern detection much more difficult.
- It allows thousands of product classes to be very loosely monitored
This is going in exactly the wrong direction. Effective post-market surveillance provides a practical solution to the safety/innovation dilemma: barriers to market entry are reduced, but risks to patient health and safety can be greatly mitigated through early warning detection. The fostering of product innovation combined with swift detection and removal of unsafe products must work in tandem: without investing in both these capabilities, the FDA is just swinging the pendulum from one set of tradeoffs to another.
Reducing barriers and increasing the speed of market entry should be balanced by more — not less — use of available RWE.
Use RWE to determine which devices should be exempted from the 510(k) process.
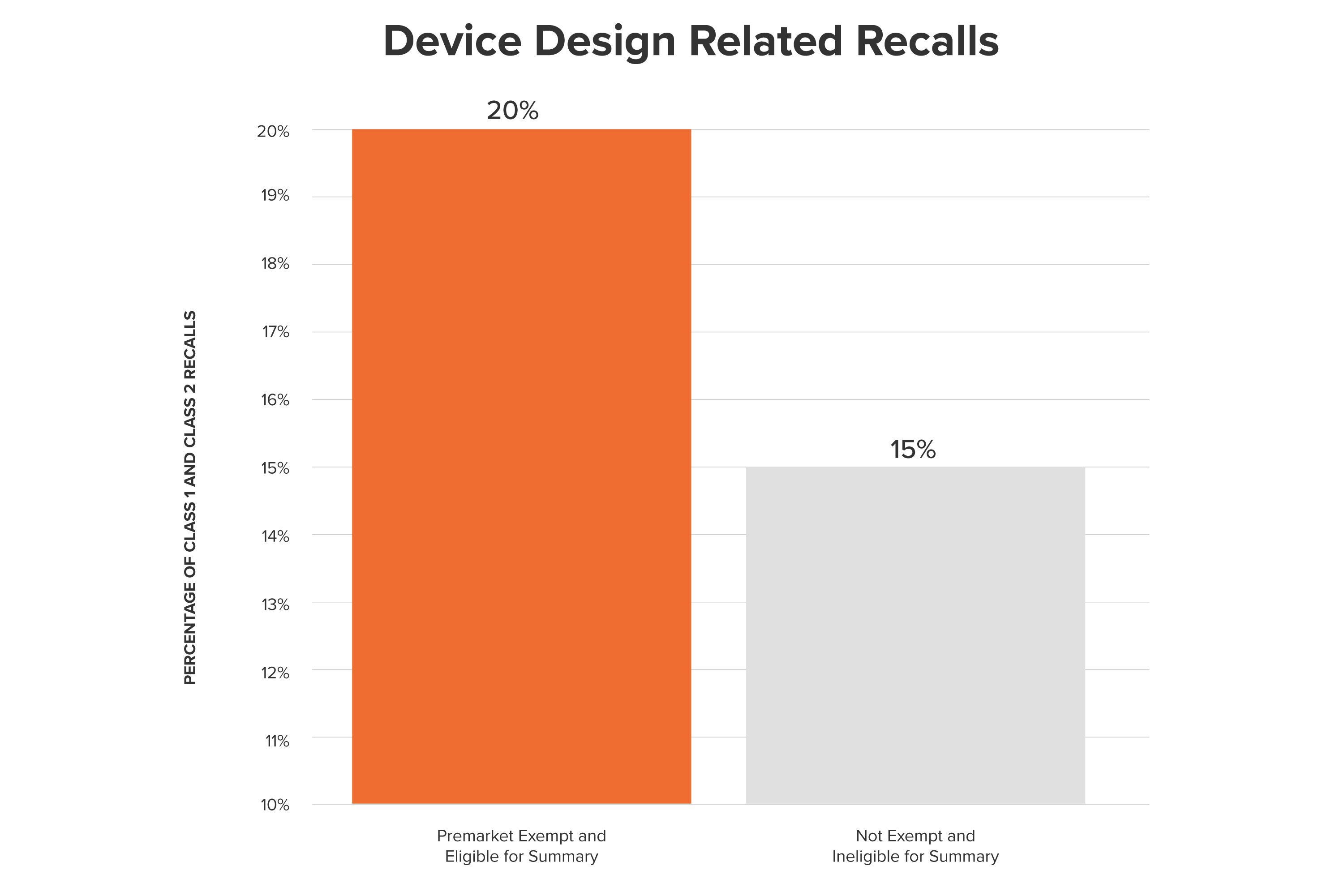
Which devices are we confident enough in to exempt from the 510(k) process? This is not a trivial question; historically, these exempted medical devices have had a mixed safety profile. Over the past 10 years, those devices are ~5% more likely to have been recalled because of device design issues than were devices that went through the 510(k) process.
In fact, while many newly exempt devices may appear to be low risk, Pharm3r’s analysis shows that some may possess serious design flaws and are linked with significant adverse events.
For example, as of January 1st, 2020, the entire class of glucose test system devices was exempted (CFR 862.1345). These devices play a key role in public health. Recently released alternative summary and MAUDE reports for this class show a high percentage of product problems, with more than 33 reports of death as a correlated outcome in the last 10 years.
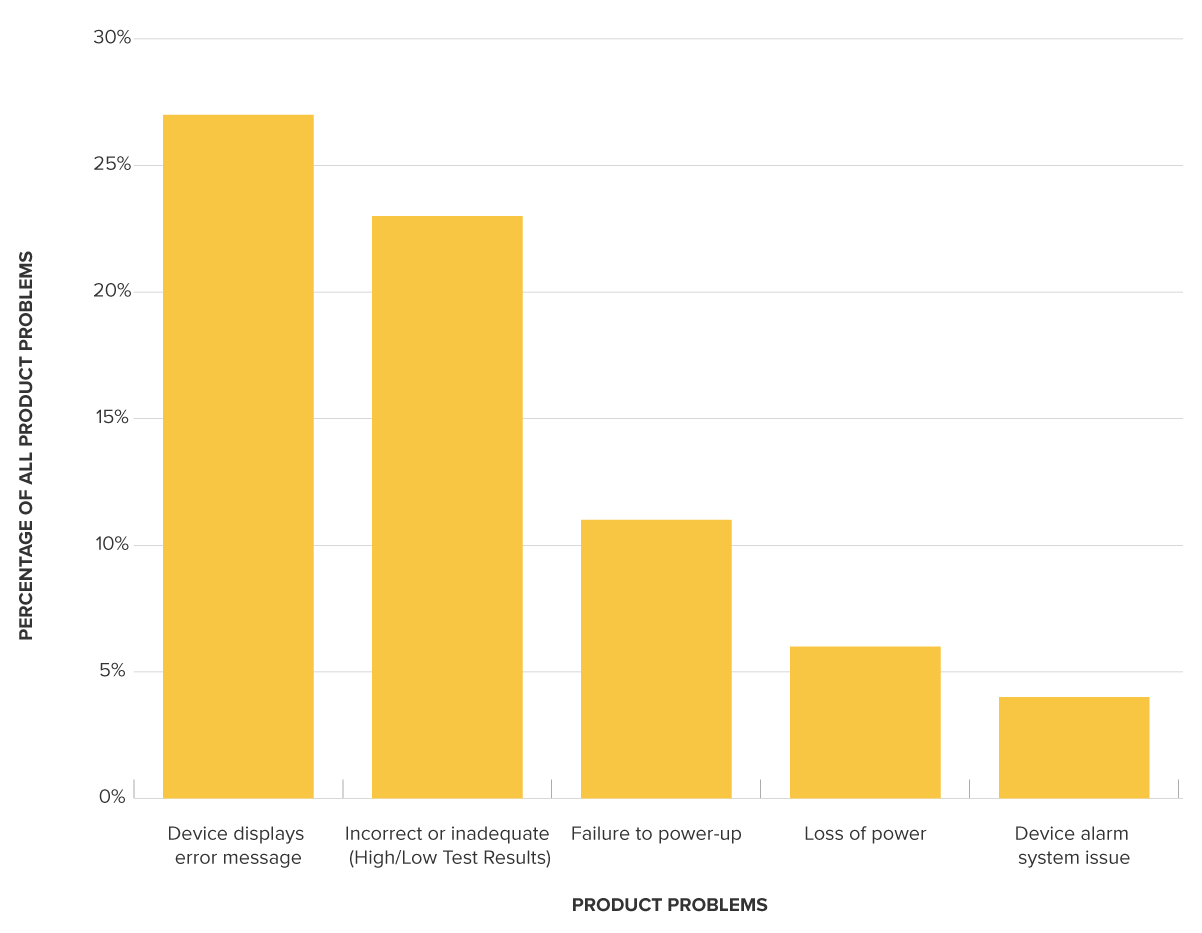
Top device problems for glucose test system class (862.1345)
There have been 60 recalls since 2008. Almost half of them were due to flaws in the design of the device, software or materials.
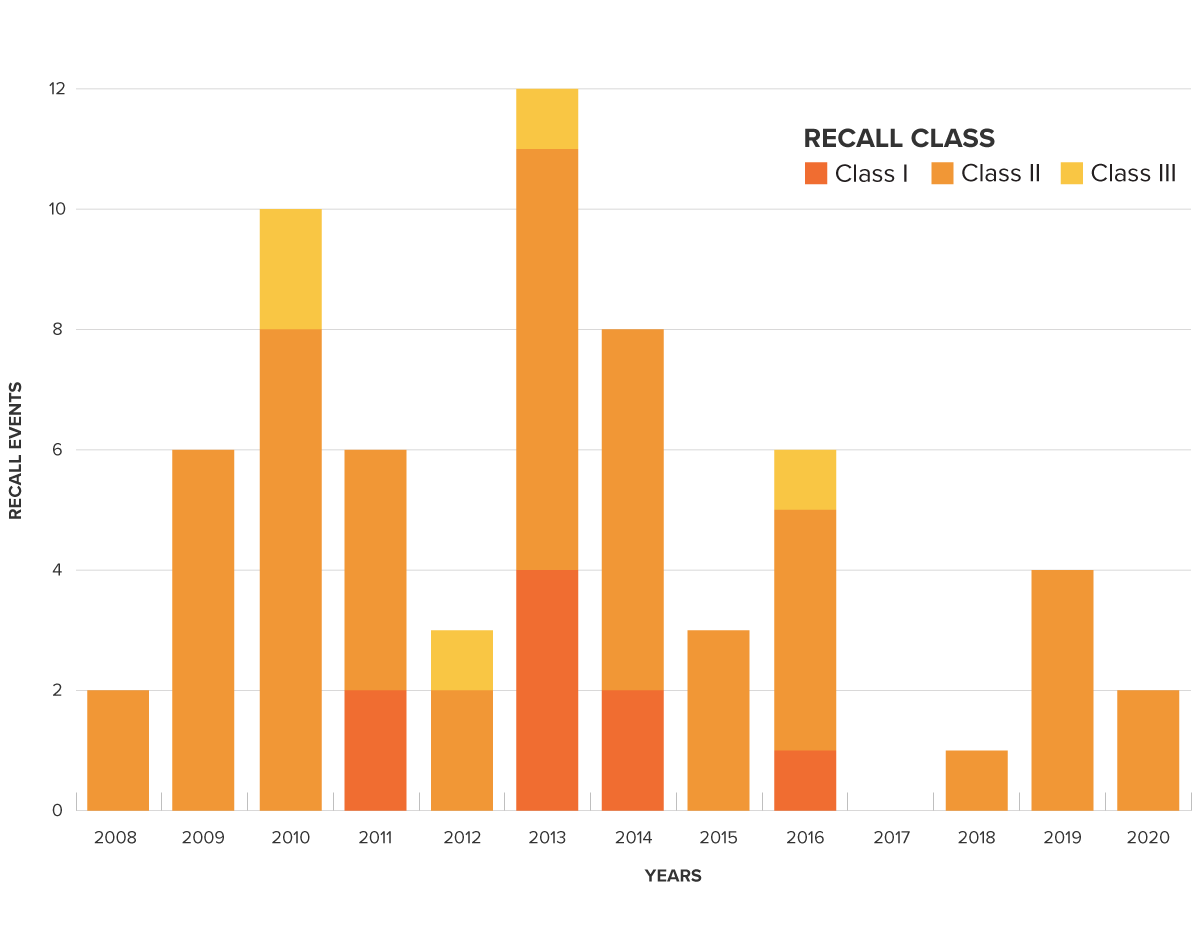
Recall Events for glucose test system class (862.1345)
A more systematic, data driven process for determining which devices are exempt from the 510(k) process would help minimize patient risk. Using an RWE approach across all devices on the market — from Class I to PMA regulated products — would better and more directly address the issue of patient risk and avoid some of these regulatory missteps, while allowing us to have fuller confidence in devices that present post-market low risk signals.
Lack of transparency of device post-market surveillance reports
No matter what the device, it is critically important that available post-market surveillance information be collected and made available in as transparent a manner as possible. Unfortunately, for many devices, this data has often been hidden in alternative databases, not filed at all, or more recently, shunted into much less informative summary versions where hundreds or thousands of patient events are summarized in one short report.
Previously hidden post market surveillance reports
The glucose monitors discussed above are a case in point. During the last 10 years over 80% of adverse event reports for this class of devices were placed in the hidden ASR database instead of the public MAUDE database. This likely led patients, health professionals, and potentially even regulators to conclude that these devices were much safer than the complete reporting record indicates.
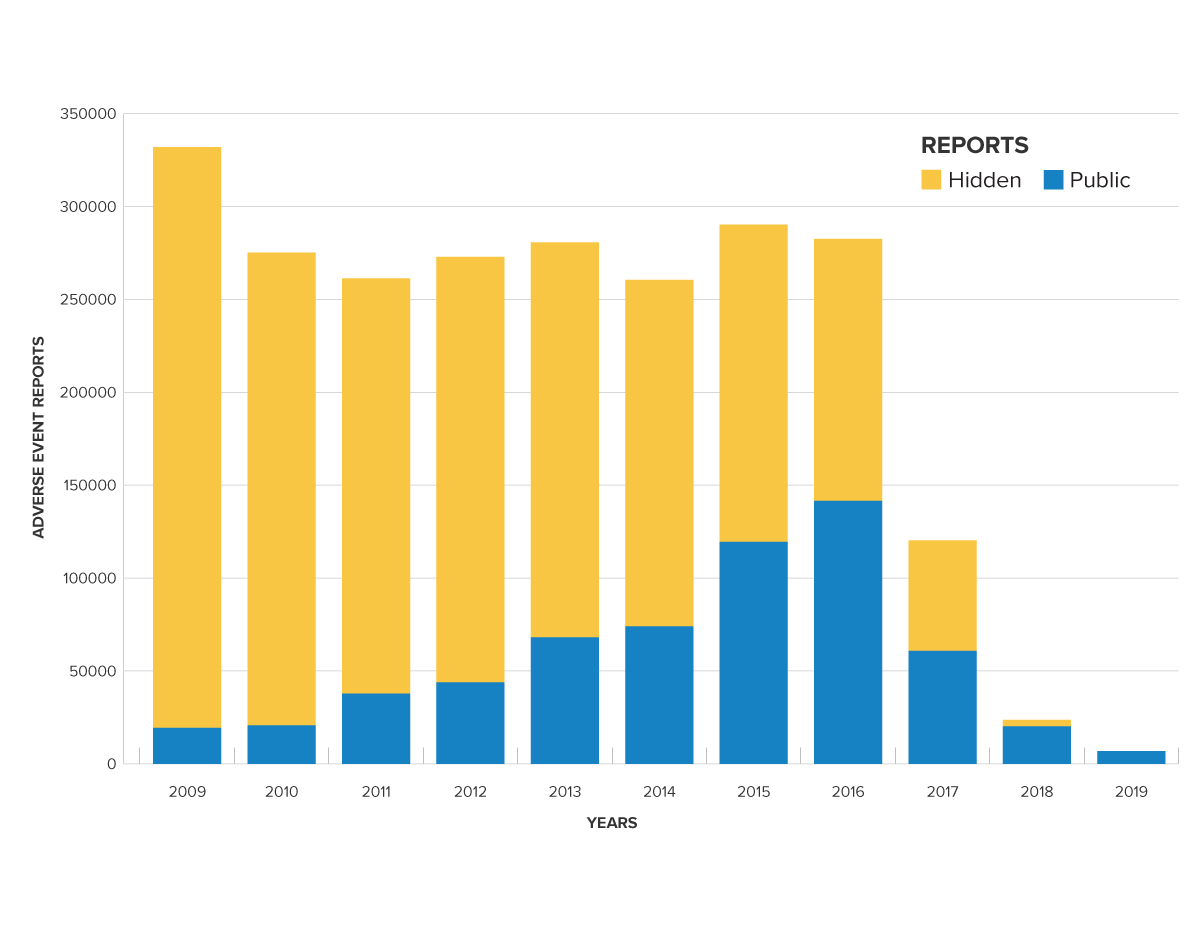
Adverse Event Reports for glucose test system class (862.1345)
Continuing disappearance of post market surveillance reports
The discontinuation of the Alternative Summary Reporting Program in April 2019 has not solved the issue of information transparency; instead, there has been an astonishing reduction of MAUDE reports for these devices. For example, one subset of over-the-counter glucose test systems (product code NBW) averaged over 200,000 reports per year in the hidden ASR database but have had virtually no MAUDE reports even after the ASR discontinuation date. While it’s theoretically possible that all design issues were suddenly solved, we think this is unlikely.
Upcoming concern: VMSR database
The Voluntary Malfunction Summary Reporting (VMSR) program was established in 2018 and permits manufacturers to report device malfunction reports in summary form on a quarterly basis. According to the FDA guidelines, this program should not apply to Class III devices and those Class II devices that are permanently implantable, life-supporting, or life-sustaining.
These restrictions do not seem to be adhered to. The FDA’s Product Classification Database indicates which product codes are eligible for summary reporting, yet in practice, we have observed cases in which ineligible devices do possess summary reports. For example, although product code OYC is ineligible for summary reporting the following screenshot was taken from the FDA Product Code Classification Database:
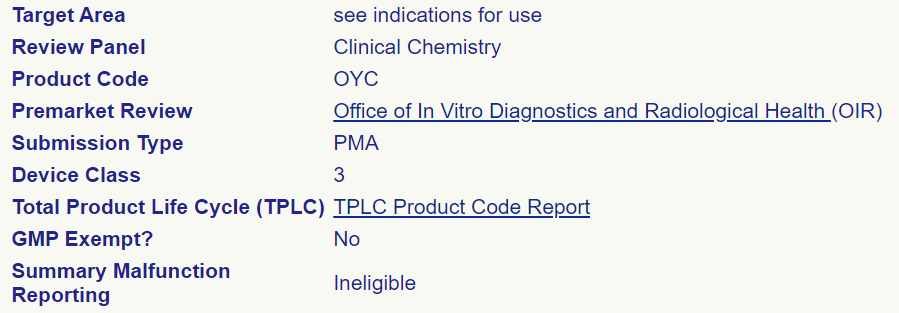
Despite this, Medtronic has submitted multiple summary reports for this class of Insulin pumps to be used with Invasive Glucose Sensor representing thousands of potential issues.
“THIS REPORT SUMMARIZES 1536 REPORTED EVENTS (1032 INITIAL REPORTS AND 504 FOLLOW-UP REPORTS). ALL REPORTED EVENTS ARE RELATED TO DEVICE MALFUNCTIONS BUTTON ERROR ALARM/KEYPAD UNRESPONSIVE AND/OR MOTOR ERROR ALARM AS ALLOWED FOR IN EXEMPTION E2015043 AND CONTAINS NO REPORTABLE SERIOUS INJURIES. PATIENT AGE RANGED FROM 5 YEARS TO 92 YEARS….”
Additionally, summary reporting also makes tracking report volume much more difficult. For example, visualization of MAUDE report volume for product code LZG (Pump, Infusion, Insulin) manufactured by Johnson and Johnson, suggests that its safety profile is improving, with fewer safety reports in recent years.
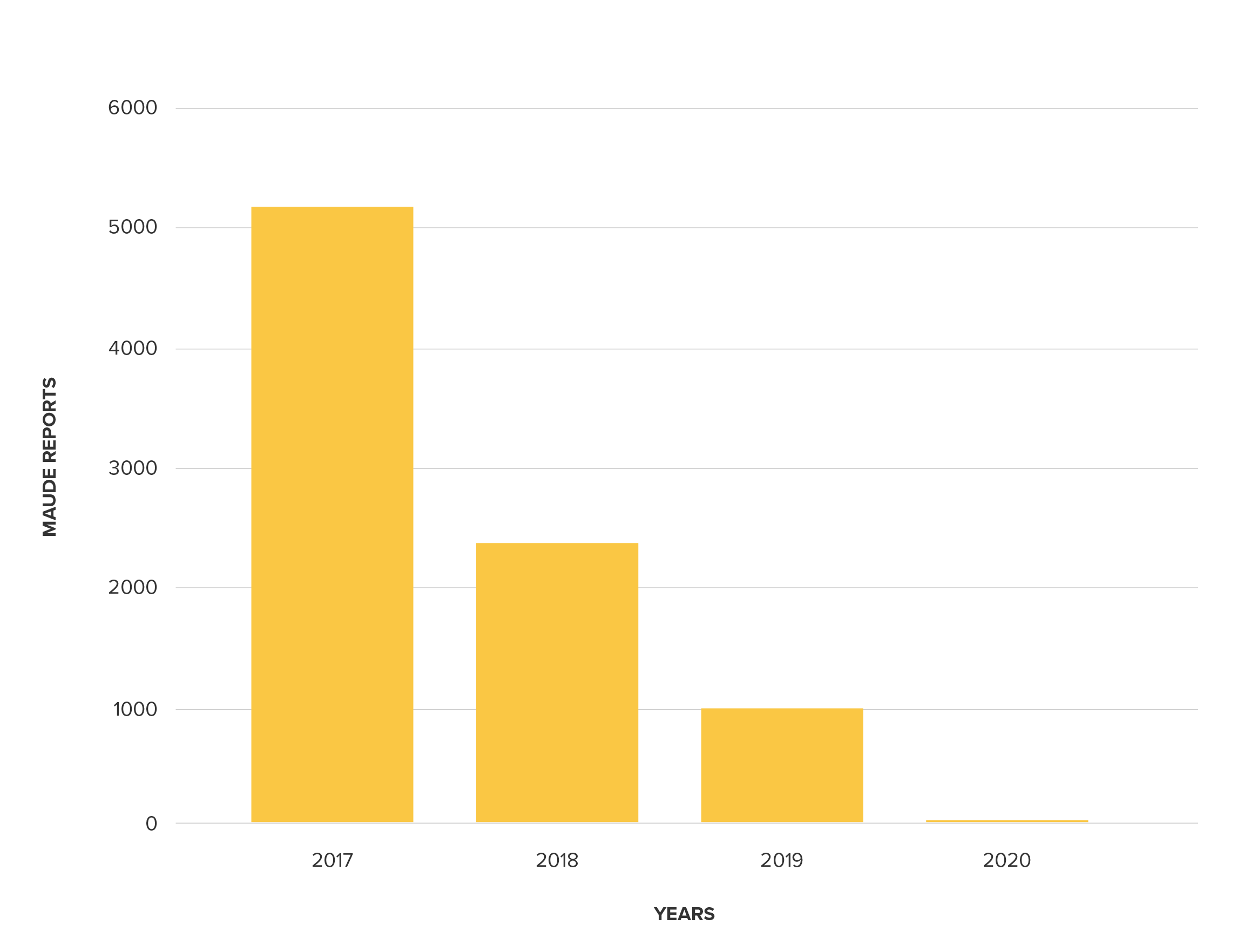
MAUDE Report trend for Johnson and Johnson LZG class products
However, in reality, 2019 saw 170 summary reports representing approximately 5,176 device malfunctions. When these buried reports are factored in, 2019 becomes the year with the highest number of reported malfunctions for code LZG. The fact that this product code is classified as ineligible for summary reporting in the first place only deepens our concern.
Pharm3r has analyzed which devices have the most missing or incomplete reports. Frankly speaking though, this arduous unearthing of buried data should not be necessary. Timely, complete, publicly available, RWE has a lot of potential to further patient safety. Sacrificing its post-market surveillance benefits by allowing less transparency puts everybody at risk.
Conclusions
Pharm3r is proud to provide an independent, real-time, evidence-based view of drug and device risk to our clients.
The FDA has an important role to play both in selecting which approval process is appropriate for which device and in mandating adequate safety reporting standards after the device has hit the market. While we very much support a more innovation-friendly posture towards product approval, our view is that the responsible way to do so is to ensure that all possible post-launch data is available at a level of granularity and detail conducive to evaluation. In this area, the FDA can and should do better.
Pharm3r’s PandoraPlus™ system, backed by its patented Boomerang NLP technology, enables real-time monitoring of over 50,000 drug and device companies and hundreds of thousands of products. Our newest software solution identifies key elements of drug and device company supply chains and pinpoints safety and geographic vulnerabilities. Contact us to learn more about our predictive risk and safety analytics.